
In zahlreichen Teams und Gruppen habe ich an unterschiedlichsten Aufgaben mitgearbeitet.
Kooperationen und Kollaboration / Initiativen, Fachgruppen, Projekte / rote, grüne, weiße Biotechnologie / Entwicklung von Plattformtechnologien, Prozessen, Leitlinien / (ultra-)Hochdurchsatz-Expression und -Screening von Enzymen und Bindern in Bakterien, Hefen und mammalischen Zellen / Immunogenität, Nutzung von Programmen und Datenbanken / …
Als Indikatoren meiner erfolgreichen und zielorientierten Arbeit kann ich hier Patente und Publikationen anführen, die aus diversen Projekten resultierten, an denen ich (zum Teil auch in leitender Funktion) beteiligt war.
Patente
Pub. No. Titel (Stichwort; Indikation; Projektart)
US2004072276A1 screening-based directed evolution (DE allgemein; intern)
WO2006125827A1 variants of serine proteases of the S1 class (DE: Änderung von Proteasen hinsichtlich Aktivität, Spezifität und Inhibition; allgemein; intern)
WO2007118889A1 specific protease for inactivation of tumour necrosis factor-alpha (DE: Abbau von TNF; Inflammation; intern)
WO2009083246A1 Antibodies to TNF Alpha (Bindung von TNF; Inflammation; intern)
WO2010148413A2, WO2011160732A1 PROTEASE VARIANTS OF HUMAN NEPRILYSIN (DE: Abbau von A-beta; Alzheimer-Krankheit; extern, MEDIMMUNE)
WO2011070088A1 ANTI-C4.4A ANTIBODIES (Bindung von C4.4A; Krebs; intern)
WO2011109452A1 Optimized Monoclonal Antibodies against Tissue Factor Pathway Inhibitor (Bindung von TFPI; Hämatologie; intern)
US2014322220A1 Anti-FGFR2 Antibodies (Bindung von FGFR2; Krebs; intern)
WO2014085527A1 HUMANIZED MONOCLONAL ANTIBODIES AGAINST ACTIVATED PROTEIN C (Bindung von aPC; Hämatologie; intern)
WO2016207090A3 TARGETED CONJUGATES OF KSP INHIBITORS (Bildung von Antikörper-Drug-Conjugaten; Krebs; intern)
WO2017042259A1 WO2018162329A1 WO2018162330A1 HPPD VARIANTS (DE: HPPD-Inhibitortoleranz; Trait Engineering; extern, CropScience)
WO2019118935A1 WO2019183150A1 NOVEL RNA-PROGRAMMABLE ENDONUCLEASE SYSTEMS AND THEIR USE IN GENOME EDITING AND OTHER APPLICATIONS (GE, DE: CRISPR-Cas endonucleases; Gentherapie; Joint Venture)
WO2019197470A1 ATRIAL NATRIURETIC PEPTIDE ENGRAFTED ANTIBODIES (Peptibody-Engineering; Herz-Kreislauf u.a.; intern)
WO2019197475A1 BRAIN NATRIURETIC PEPTIDE ENGRAFTED ANTIBODIES (Peptibody-Engineering; Herz-Kreislauf u.a; intern)
WO2019197477A1 C-TYPE NATRIURETIC PEPTIDE ENGRAFTED ANTIBODIES (Peptibody-Engineering; Herz-Kreislauf u.a.; intern)
WO2022180145A2 INHIBITORS OF IL-11 OR IL-11RA FOR USE IN THE TREATMENT OF ABNORMAL UTERINE BLEEDING (Bindung von IL-11; Frauengesundheit; intern)
WO2023168432A2 HUMAN ANTIBODIES AGAINST ACTIVATED PROTEIN C AND USES THEREOF (Bindung von aPC; Hämatologie; extern)
Publikationen
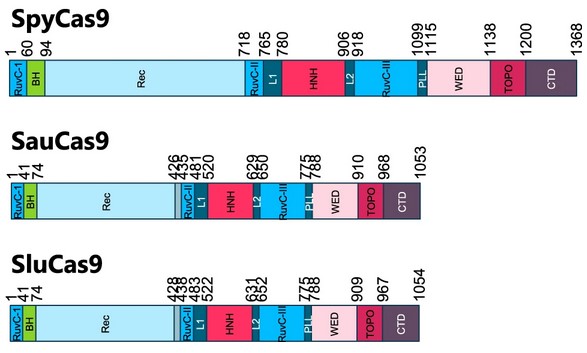
Improved CRISPR genome editing using small highly active and specific engineered RNA-guided nucleases.
2021, Nature Communications
Streptococcus pyogenes (Spy) Cas9 has potential as a component of gene therapeutics for incurable diseases. One of its limitations is its large size, which impedes its formulation and delivery in therapeutic applications. Smaller Cas9s are an alternative, but lack robust activity or specificity and frequently recognize longer PAMs. Here, we investigated four uncharacterized, smaller Cas9s and found three employing a “GG” dinucleotide PAM similar to SpyCas9. Protein engineering generated synthetic RNA-guided nucleases (sRGNs) with editing efficiencies and specificities exceeding even SpyCas9 in vitro and in human cell lines on disease-relevant targets. sRGN mRNA lipid nanoparticles displayed manufacturing advantages and high in vivo editing efficiency in the mouse liver. Finally, sRGNs, but not SpyCas9, could be packaged into all-in-one AAV particles with a gRNA and effected robust in vivo editing of non-human primate (NHP) retina photoreceptors. Human gene therapy efforts are expected to benefit from these improved alternatives to existing CRISPR nucleases. See publication
Targeted inhibition of activated protein C by a non-active-site inhibitory antibody to treat hemophilia.
2020, Nature Communications
Activated protein C (APC) is a plasma serine protease with antithrombotic and cytoprotective functions. Based on the hypothesis that specific inhibition of APC’s anticoagulant but not its cytoprotective activity can be beneficial for hemophilia therapy, 2 types of inhibitory monoclonal antibodies (mAbs) are tested: A type I active-site binding mAb and a type II mAb binding to an exosite on APC (required for anticoagulant activity) as shown by X-ray crystallography. Both mAbs increase thrombin generation and promote plasma clotting. Type I blocks all APC activities, whereas type II preserves APC’s cytoprotective function. In normal monkeys, type I causes many adverse effects including animal death. In contrast, type II is well-tolerated in normal monkeys and shows both acute and prophylactic dose-dependent efficacy in hemophilic monkeys. Our data show that the type II mAb can specifically inhibit APC’s anticoagulant function without compromising its cytoprotective function and offers superior therapeutic opportunities for hemophilia. See publication
Engineering Potent, Small, Chimeric, Synthetic, RNA-Guided Nucleases (sRGN) from Four Uncharacterized Cas9 Genes
2019 ASGCT Annual – Meeting Abstract No. 135, Measuring and Mitigating Genotoxicity of Genome Editing
Adeno-associated viruses (AAVs) and lipid nanoparticles (LNP) are among the methods of choice for delivery of nucleases for in-vivo genome editing. The widely used and highly active Streptococcus pyogenes (Spy) Cas9 nuclease is specific for an advantageously short and non-degenerate PAM sequence. However, the large size of SpyCas9, in combination with required sgRNA and expression elements, presents a challenge to the 4.5 kb DNA packaging limit of AAVs, as well as to the synthesis of long mRNA templates and their stable formulation into LNPs. On the other hand, the best-characterized smaller Cas9s, such as Staphylococcus aureus Cas9, frequently recognize degenerate and longer PAMs, which significantly reduce the number of addressable genomic target sites. To address these issues, we evaluated four related, previously uncharacterized Cas9 nucleases of ~1050 amino acids in length. Surprisingly, most were found to be specific for a common, non-degenerate, small PAM motif. Using multiple protein engineering approaches, we altered these genes to generate novel, chimeric, synthetic, RNA-guided nucleases (sRGNs) and demonstrated efficient editing in human cells. Analyses using all 60 possible, single-nucleotide mismatched off-targets of a DNA substrate indicated high overall specificity for each clone. Importantly, the different clones displayed a localized signature of higher specificity for different mismatches at different sites along the target. Thus, we have generated a set of novel, small sRGNs, from which improved nucleases can be selected for particular targets of interest. LNP packaging and in-vivo performance vs. SpyCas9 will be reported in a separate abstract. Because these sRGNs were not derived from Spy or S. aureus Cas9s, we are working toward assessing these alternative nucleases regarding the potential for lower occurrence of pre-existing antibodies in human populations. All in all, these engineered, chimeric, synthetic, RNA-guided nucleases are thus expected to be valuable additions to the canon of known genome-editing nucleases that can be employed for human gene therapy applications. See publication
Preclinical Antitumor Efficacy of BAY 1129980—a Novel Auristatin-Based Anti-C4.4A (LYPD3) Antibody–Drug Conjugate for the Treatment of Non–Small Cell Lung Cancer
2017, American Association for Cancer Research – Mol Cancer Therapeutics
C4.4A (LYPD3) has been identified as a cancer- and metastasis-associated internalizing cell surface protein that is expressed in non–small cell lung cancer (NSCLC), with particularly high prevalence in the squamous cell carcinoma (SCC) subtype. With the exception of skin keratinocytes and esophageal endothelial cells, C4.4A expression is scarce in normal tissues, presenting an opportunity to selectively treat cancers with a C4.4A-directed antibody–drug conjugate (ADC). We have generated BAY 1129980 (C4.4A-ADC), an ADC consisting of a fully human C4.4A-targeting mAb conjugated to a novel, highly potent derivative of the microtubule-disrupting cytotoxic drug auristatin via a noncleavable alkyl hydrazide linker. In vitro, C4.4A-ADC demonstrated potent antiproliferative efficacy in cell lines endogenously expressing C4.4A and inhibited proliferation of C4.4A-transfected A549 lung cancer cells showing selectivity compared with a nontargeted control ADC. In vivo, C4.4A-ADC was efficacious in human NSCLC cell line (NCI-H292 and NCI-H322) and patient-derived xenograft (PDX) models (Lu7064, Lu7126, Lu7433, and Lu7466). C4.4A expression level correlated with in vivo efficacy, the most responsive being the models with C4.4A expression in over 50% of the cells. In the NCI-H292 NSCLC model, C4.4A-ADC demonstrated equal or superior efficacy compared to cisplatin, paclitaxel, and vinorelbine. Furthermore, an additive antitumor efficacy in combination with cisplatin was observed. Finally, a repeated dosing with C4.4A-ADC was well tolerated without changing the sensitivity to the treatment. Taken together, C4.4A-ADC is a promising therapeutic candidate for the treatment of NSCLC and other cancers expressing C4.4A. A phase I study (NCT02134197) with the C4.4A-ADC BAY 1129980 is currently ongoing. See publication
Characterization of a High-Affinity Fully Human IgG2 Antibody Against Tissue Factor Pathway Inhibitor As a Bypass Agent for the Treatment of Hemophilia
2015, American Society of Hematology – Blood – Meeting Abstract
The tissue factor initiated coagulation pathway is intact in patients with hemophilia; however, this pathway is normally inhibited by tissue factor pathway inhibitor (TFPI), resulting in insufficient thrombin generation to stop bleeding and prevent recurrence of bleeding. Antagonizing this natural inhibition through anti-TFPI antibodies is a potential mechanism to effectively restore hemostasis in patients with hemophilia. A human phage-displayed antibody library was used to identify optimal anti-TFPI antibodies. From the panning campaign, unique antibodies were identified that bound to human TFPI and were cross-reactive to murine TFPI. In vitro characterization demonstrated that 6 of the antibodies could block TFPI activity, partially or completely restoring factor Xa activity. These 6 antibodies also shortened clotting time of hemophilic human (Hem A) plasma in a diluted prothrombin assay. When the ROTEM assay (Tem International GmbH, Basel, Switzerland) was used to measure clotting in human Hem A plasma from congenitally deficient individuals (factor VIII [FVIII] levels <0.38%), anti-TFPI antibodies significantly shortened clotting time, alone or in combination with FVIII or activated factor VII. In addition, the antibodies were effective in shortening clotting time in human blood containing anti-FVIII antibodies, indicating that anti-TFPI antibodies can be applied to inhibitor patients. One of the identified anti-TFPI antibodies was tested in a hemophilia A mice tail vein transection model and improved survival rate to 60%, significantly higher than the 10% survival rate of mice treated with an isotype control antibody. Additionally, combined with low-dose FVIII, this antibody could further increase the survival rate in hemophilic mice, and it was selected for further optimization. Optimization covered 2 different aspects: (1) affinity maturation to increase its affinity to human and murine TFPI; and (2) sequence optimization to reduce sequence deviation from germ-line sequences and to remove critical residues prone to undergo unwanted chemical modifications during production and/or storage. Both processes consisted of 2 rounds of sequence optimization and screening. Following affinity maturation, target affinity was increased by >200-fold on human and >500-fold on murine TFPI. Sequence optimization was performed on the backbone of the final affinity-matured variant. BAY 1093884, the final optimized variant, is a fully human IgG2 antibody with <10 pM binding affinity to human and murine TFPI. See publication
Sustained peripheral depletion of amyloid-β with a novel form of neprilysin does not affect central levels of amyloid-β
2014, Oxford Academic Journals – Brain
Alzheimer’s disease is characterized by the accumulation of amyloid deposits in the brain and the progressive loss of cognitive functions. Although the precise role of amyloid-β in disease progression remains somewhat controversial, many efforts to halt or reverse disease progression have focussed on reducing its synthesis or enhancing its removal. It is believed that brain and peripheral soluble amyloid-β are in equilibrium and it has previously been hypothesized that a reduction in peripheral amyloid-β can lower brain amyloid-β, thereby reducing formation of plaques predominantly composed of insoluble amyloid-β; the so-called peripheral sink hypothesis. Here we describe the use of an amyloid-β degrading enzyme, the endogenous metallopeptidase neprilysin, which is fused to albumin to extend plasma half-life and has been engineered to confer increased amyloid-β degradation activity. We used this molecule to investigate the effect of degradation of peripheral amyloid-β on amyloid-β levels in the brain and cerebrospinal fluid after repeated intravenous dosing for up to 4 months in Tg2576 transgenic mice, and 1 month in rats and monkeys. This molecule proved highly effective at degradation of amyloid-β in the periphery but did not alter brain or cerebrospinal fluid amyloid-β levels, suggesting that the peripheral sink hypothesis is not valid and is the first time that this has been demonstrated in non-human primates. See publication
Engineering Neprilysin Activity and Specificity to Create a Novel Therapeutic for Alzheimer’s Disease
2014, PLOS JOURNALS – PLOS ONE
Neprilysin is a transmembrane zinc metallopeptidase that degrades a wide range of peptide substrates. It has received attention as a potential therapy for Alzheimer’s disease due to its ability to degrade the peptide amyloid beta. However, its broad range of peptide substrates has the potential to limit its therapeutic use due to degradation of additional peptides substrates that tightly regulate many physiological processes. We sought to generate a soluble version of the ectodomain of neprilysin with improved activity and specificity towards amyloid beta as a potential therapeutic for Alzheimer’s disease. Extensive amino acid substitutions were performed at positions surrounding the active site and inner surface of the enzyme and variants screened for activity on amyloid beta 1–40, 1–42 and a variety of other physiologically relevant peptides. We identified several mutations that modulated and improved both enzyme selectivity and intrinsic activity. Neprilysin variant G399V/G714K displayed an approximately 20-fold improved activity on amyloid beta 1–40 and up to a 3,200-fold reduction in activity on other peptides. Along with the altered peptide substrate specificity, the mutant enzyme produced a markedly altered series of amyloid beta cleavage products compared to the wild-type enzyme. Crystallisation of the mutant enzyme revealed that the amino acid substitutions result in alteration of the shape and size of the pocket containing the active site compared to the wild-type enzyme. The mutant enzyme offers the potential for the more efficient degradation of amyloid beta in vivo as a therapeutic for the treatment of Alzheimer’s disease. See publication
Purine nucleoside phosphorylase from Cellulomonas sp.: Physicochemical properties and binding of substrates determined by ligand-dependent enhancement of enzyme intrinsic fluorescence, and by protective effects of ligands on thermal inactivation of the enzyme
2002, Elsevier – Biochimica et Biophysica Acta – Protein Structure and Molecular Enzymology
Purine nucleoside phosphorylase (PNP) from Cellulomonas sp., homotrimeric in the crystalline state, is also a trimer in solution. Other features of the enzyme are typical for “low molecular mass” PNPs, except for its unusual stability at pH 11. Purine bases, α-d-ribose-1-phosphate (R1P) and phosphate enhance the intrinsic fluorescence of Cellulomonas PNP, and hence form binary complexes and induce conformational changes of the protein that alter the microenvironment of tryptophan residue(s). The effect due to guanine (Gua) binding is much higher than those caused by other ligands, suggesting that the enzyme preferentially binds a fluorescent, most probably rare tautomeric anionic form of Gua, further shown by comparison of emission properties of the PNP/Gua complex with that of Gua anion and its N-methyl derivatives. Guanosine (Guo) and inosine (Ino) at 100 μM concentration show little and no effect, respectively, on enzyme intrinsic fluorescence, but their protective effect against thermal inactivation of the enzyme points to their forming weak binary complexes with PNP. Binding of Gua, hypoxanthine (Hx) and R1P to the trimeric enzyme is described by one dissociation constant, Kd=0.46 μM for Gua, 3.0 μM for Hx, and 60 μM for R1P. The binding stoichiometry for Gua (and probably Hx) is three ligand molecules per enzyme trimer. Effects of phosphate on the enzyme intrinsic fluorescence are due not only to binding, but also to an increase in ionic strength, as shown by titration with KCl. When corrected for effects of ionic strength, titration data with phosphate are most consistent with one dissociation constant, Kd=270 μM, but existence of a very weak binding site with Kd>50 mM could not be unequivocally ruled out. Binding of Gua to the PNP/phosphate binary complex is weaker (Kd=1.7 μM) than to the free enzyme (Kd=0.46 μM), suggesting that phosphate helps release the purine base in the catalytic process of phosphorolysis. The results indicate that nonlinear kinetic plots of initial velocity, typical for PNPs, including Cellulomonas PNP, are not, as generally assumed, due to cooperative interaction between monomers forming the trimer, but to a more complex kinetic mechanism than hitherto considered. See publication
Crystallization and preliminary X-ray analyses of catabolite control protein A, free and in complex with its DNA-binding site
2000, IUCr Journals – Acta Crystallographica Section D
The catabolite control protein (CcpA) from Bacillus megaterium is a member of the bacterial repressor protein family GalR/LacI. CcpA with an N-terminal His-tag was used for crystallization. Crystals of free CcpA and of CcpA in complex with the putative operator sequence (catabolite responsive elements, CRE) were obtained by vapour-diffusion techniques at 291 K using the hanging-drop method. CcpA crystals grown in the presence of polyethylene glycol 8000 belong to the hexagonal space group P6122 or P6522, with unit-cell parameters a = 74.4, c = 238.8 Å. These crystals diffract X-rays to 2.55 Å resolution and contain one monomer of the homodimeric protein per asymmetric unit. Crystals of the CcpA-CRE complex were obtained with ammonium sulfate as precipitant and belong to the tetragonal space group I4122, with unit-cell parameters a = 125, c = 400 Å and one complex per asymmetric unit. Although these co-crystals grew to a sufficient size, X-ray diffraction was limited to 8 Å resolution. See publication
Binding of substrates by purine nucleoside phosphorylase (PNP) from Cellulomonas sp. – Kinetic and spectrofluorimetric studies
1999, Elsevier – Nucleosides and Nucleotides
Dissociation constants and stoichiometry of binding for interaction of Cellulomonas sp. purine nucleoside phosphorylase with its substrates: inosine/guanosine, orthophosphate, guanine/hypoxanthine and D-ribose-1-phosphate were studied by kinetic and spectrofluorimetric methods. See publication
Crystal structure of the purine nucleoside phosphorylase (PNP) from Cellulomonas sp. and its implication for the mechanism of trimeric PNPs
1999, Elsevier – Journal of Molecular Biology
The three-dimensional structure of the trimeric purine nucleoside phosphorylase (PNP) from Cellulomonas sp. has been determined by X-ray crystallography. The binary complex of the enzyme with orthophosphate was crystallized in the orthorhombic space group P212121 with unit cell dimensions a = 64.1 Å, b = 108.9 Å, c = 119.3 Å and an enzymatically active trimer in the asymmetric unit. X-ray data were collected at 4 °C using synchrotron radiation (EMBL/DESY, Hamburg). The structure was solved by molecular replacement, with the calf spleen PNP structure as a model, and refined at 2.2 Å resolution. The ternary “dead-end” complex of the enzyme with orthophosphate and 8-iodoguanine was obtained by soaking crystals of the binary orthophosphate complex with the very weak substrate 8-iodoguanosine. Data were collected at 100 K with CuKα radiation, and the three-dimensional structure refined at 2.4 Å resolution. See publication
Cellulomonas sp. purine nucleoside phosphorylase (PNP): Comparison with human and E. coli enzymes
1998, Springer – Advances in Experimental Medicine and Biology
The ubiquitous enzyme purine nucleoside phosphorylase (PNP, E.C. 2.4.2.1.) catalyzes the reversible phosphorolysis of naturally occurring purine nucleosides, and many analogues, as follows:
β−purinenucleoside+orthophosphate⇔purine+α−D−pentose−1−phosphate
Inhibitors of PNP are potential immunosuppressive, antiviral and antiparasitic agents. Properties of PNP from various sources are also of interest in light of reports that the product of the E. coli DeoD (PNP) gene mediates the killing of melanoma cells.
Two main classes of PNPs have been characterized, so-called „low-molecular weight“ (M, ~90 kDa), trimeric, mainly mammalian enzymes, specific for 6-oxo-purines (e.g. human erythrocytes and calf spleen), and less specific „high-molecular weight“ (M, ~110-140 kDa), mainly hexameric, bacterial, phosphorylases (e.g. E. coli., S. typhimurium) that accept as substrates 6-oxo-and 6-aminopurines, as well as some unusual purine analogues like benzimidazole. We herein describe some of the properties of the PNP from Cellulomonas sp. which differs from the foregoing in that some of its properties are common to both classes. See publication
Crystallization and preliminary X-ray studies of purine nucleoside phosphorylase from Cellulomonas sp.
1998, IUCr Journals – Acta Crystallographica Section D
The commercially available enzyme purine nucleoside phosphorylase (PNP) from Cellulomonas sp. was purified by ion-exchange chromatography, partially sequenced and crystallized in two different crystal forms using the hanging-drop vapour-diffusion technique. Crystal form A grows as polyeders and/or cubes in the cubic space group P4232 with unit-cell dimension a = 162.5 Å. Crystal form B appears as thick plates in the space group P212121 with unit-cell dimensions a = 63.2, b = 108.3 and c = 117.4 Å. Both crystal forms contain three monomers (one trimer) in the asymmetric unit. See publication